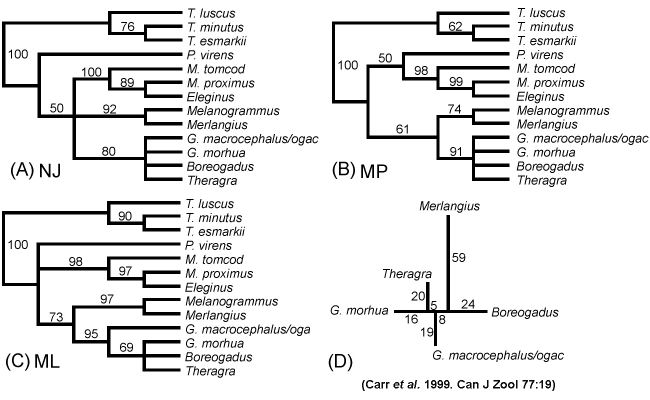
Neighbor-Joining (A), Maximum Parsimony (B), and Maximum Likelihood (C) cladograms, with bootstrap support; parsimony phylogram, with branch lengths (D)
(B) Cladistics: given the distribution of various character states (A,C,G,T), which branching order requires the smallest number of changes? [e.g., MP: Maximum Parsimony]
(C) Maximum-Likelihood:
Given a particular set of parameters
for a model of (molecular) evolution,
which of the many unlikely outcomes is least unlikely (maximally
likely)?
(D) Cladograms
vs Phylograms:
diagrams of evolutionary relationships may be cladograms that simply depict branching
order (A, B, & C), or phylograms that show the amount of
change occuring along each branch (D)
Bootstrap Analysis: Given
a finite data set, re-sample it as though it were a random sample of an
infinite data set. In what % of cases are the same groups observed?
[(A-C) numbers above branches, applicable to groups to the right]